jump to exam page:
Recent comments (see more)
...
∗niboonsh made a comment
(
nbme21)
d_holles
Amazing video dude. Somehow never learned this in neuro lol.
+7
aag
Awesome video! Is this why you can give Mg2+ to eclampsia patients, because if so, mind goddamn blown.
+4
...
∗drdoom made a comment
(
nbme21)
qball
Awesome explanation. Now explain it to me like I'm 5.
+13
drdoom
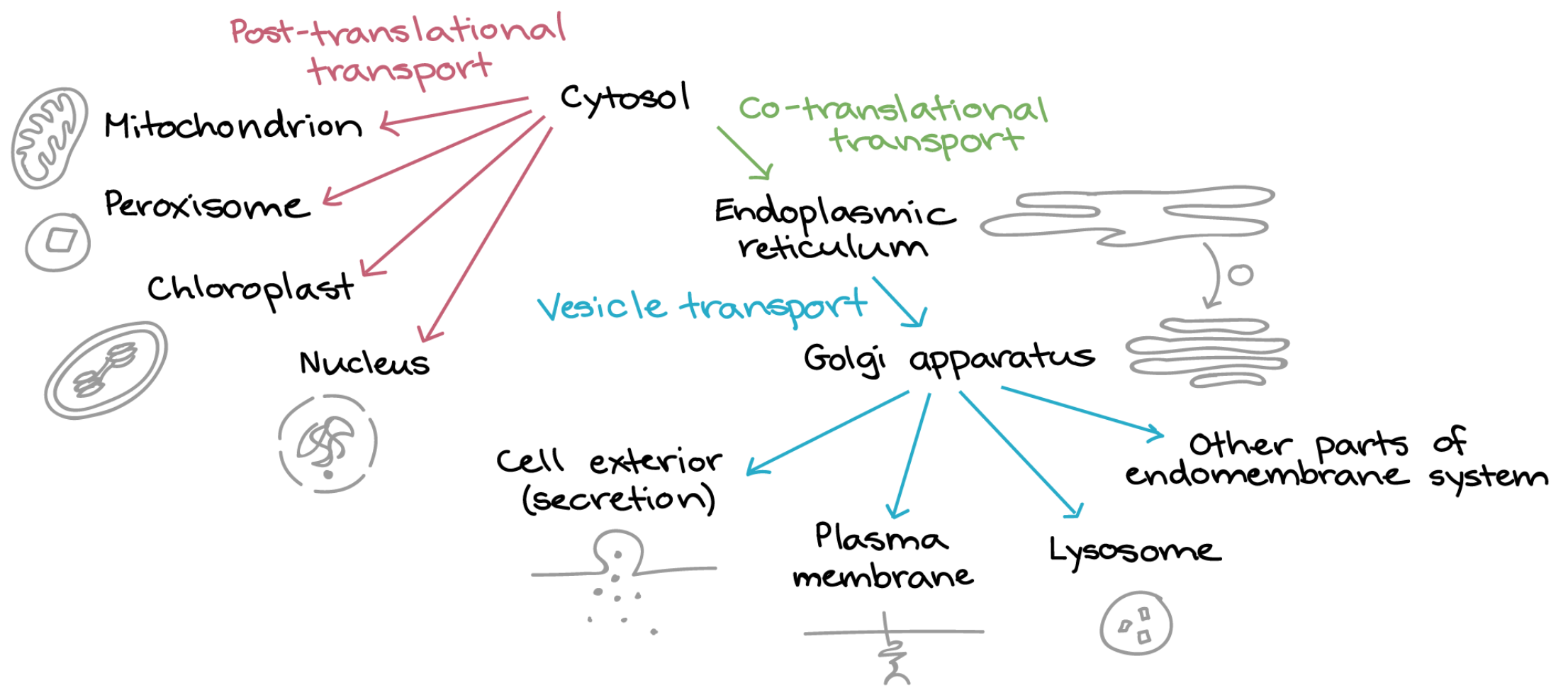
All baby peptides are born in the cytosol. But some baby peptides have a birthmark at their N-terminus. The birthmark tells a special mailman that this baby needs to be delivered somewhere else. If you chop off the birthmark — or erase it somehow — the mailman never knows to take baby to its true home. The end. Now go to sleep or Santa won’t bring your presents.
+83
...
∗drdoom made a comment
(
nbme24)
regularstudent
Isn't the brush border still part of the intestinal lumen? Don't the amino acids enter into the intestinal cell (the "intestinal mucosa")?
+1
drdoom
@regularstudent No, the lumen is literally the cavity—the empty space.
+2
drdoom
@jj375 best schematic i’ve seen so far. superb.
+2
...
∗seagull made a comment
(
nbme22)
aesalmon
I agree, I picked H1 because such a common complaint for those on TCAs is Sedation, I figure it might be so commonly seen as to be the "most common" reason for noncompliance. I suppose the "hot as a hare...etc" effects would be more severe/annoying, but I didn't think they were more common.
+4
fcambridge
I just like to pretend that there's a reason this question is now in an NBME and no longer being used for the test. Hopefully they realized the idiocy of this question like we all do
+1
link981
Since it said cyclic, I thought of using, discontinuing, then using again. These people who write these questions need take some English writing courses so they can write with CLARITY. Cyclic is not the same as Tricyclic.
+6
waterloo
Incredibly awful question. one thought I did have when deciding between anticholinergic and antihistaminic - nortriptyline and desipramine are secondary amines that have less anti-cholinergic effects (from Sketchy Pharm) so maybe that's what they were getting at? That someone went out and made a new TCA drug that would have less anticholinergic effects.
+
victor_abdullatif
This isn't testing drug epidemiology; it's actually asking "which of these side effects are caused by TCAs and would be the worst to experience?"
+
tekkenman101
"worst to experience" is incredibly subjective lmao.
+1
...
∗azibird made a comment
(
nbme24)
yousif11
This question did not age well
+23
helppls
I'm going to be taking Step with this song stuck in my head lol
+
...
∗lsmarshall made a comment
(
nbme24)
lsmarshall
Summary of metabolic issues relating to hyperammonemia
+9
seagull
i'm leaning towards Ornithine transcarbamylase deficiency.
+3
notadoctor
Not sure why this isn't considered a mitochondrial disorder since the issue is Ornithine transcarbamylase deficiency in the mitochondria?
+2
wowo
figure in OTC deficiency, they might have to explicitly mention the orotic aciduria AND typically presents earlier, around 24-48hrs of life after they've fed (at least per BB)
+ also per BB, propionic acidemia and MM acidemia have an onset of weeks to months and lead to build up of organic acids --> acidemia in addition to hyperammonemia (not sure why, but several aa enter the TCA cycle via propionyl CoA --> methylmalonyl CoA --> succinyl CoA, but now this is defunct d/t enzyme deficiencies...?). Anywho, propionic acidemia described on FA2019 p85, but doesn't list hyperammonemia
+2
artist90
i think it cannot be Ornithine transcarbamylase deficiency bc it is XR disease. this pt has a healthy 2yr old brother which rules out X-linked recessive disease correct me if i m wrong
+7
artist90
it is 100% Propionic acidemia Uworld Q-id: 1340. it is an exact copy question of uworld. i got it wrong bc i forgot these are organic acids. But i am still confused on 2 things
1-how does acidosis cause Hypoglycemia and Ketosis.
2-why is Ammonia elevated in these pts bc urea cycle will be fine?
+2
yb_26
1) hyperammonemia is seen in all urea cycle disorders except arginase deficiency
2) organic acids directly inhibit urea cycle => hyperammonemia (from UWorld)
+2
yex
According to UW, there is another question # 1341. This one refers to methylmalonic acidemia (ORGANIC ACIDEMIA). HYPOGLYCEMIA results from overall increased metabolic rate leading to increased glucose utilization and direct toxic (-) of gluconeogenesis by organic acids. The presence of hypoglycemia leads to increased free fatty acid metabolism that produces KETONES, resulting in a further anion gap met acidocis. Finally, organic acids also directly (-) the urea cycle, leading to HYPERAMMONEMIA.
+19
ih8payingfordis
I think there has been some confusions because FA does a poor job integrating these concepts together but here are some references:
FA 2019 pg 85: Propionic Acidemia (this is one example of organic acid metabolism disorder) In the summary figure on Pg 74, look at the protein metabolism (located bottom right). Basically, you can have disorders where you have Propionyl-CoA buildup alone OR Proionyl-CoA and Methylmalonyl-CoA buildup.
And these organic acidemias are autosomal recessive in their inheritance pattersn, which fit with the stem we're given (ie XX individuals can be affected)
One thing that UW state that I do not have an explanation for: the buildup of organic acids inhibit the urea cycle hence the increase in ammonia (anyone knows how this happens?)
+
jaramaiha
@aaftabsethi because all of the siblings would show some symptoms if it were to be a mitochondrial disease. Which his brother is healthy, and sister was full-term but died due to unspecified causes.
+
utap2001
Why acidosis cause Hypoglycemia and Ketosis and hyperammonia? In acidosis(such lactic acidosis)-> indicate body energy crisis -> inc. glycolysis, Hypoglycemia-> inc. protein breakdown, Hyperammonia -> dec. TCA cycle, Ketosis
+
...
∗tea-cats-biscuits made a comment
(
nbme24)
atstillisafraud
Thanks for a good answer. This question made me feel like I was taking T21 pills
+18
medguru2295
Thank you- I was really thinking this question had 2 correct answers... of course my dumbass picked Mast cells.
+7
ajss
where do i find this info??
+
paperbackwriter
@ajss pg 112 of first aid 2019, under type I hypersensitivity. Immediate --> mast cells releasing histamine and tryptase, late--> eosinophils and leukotrienes recruited via chemokines
+3
graciewacie9
Wow, i missed the fact that the question is asking for the RESULT of the reaction, NOT the cause of the reaction. Mast cells cause the initial reaction, eosinophils would be the result of the eosinophils. *facepalm
+9
lba9587
Pathophys (as far as I understand it)...Mast cell degranulates, thus the phospholipid bilayer et. Al are left behind and needs to be degraded. Who comes in? Our good friend eosinophils, as they contain Major Basic Protein (responsible from breakdown of expired mast cell).
Note, you can tie this in to the delayed Leukotriene effects of an allergic rxn, as the bilayer is also broken down by arach. Acid.
(See this link to support my credibility https://images.app.goo.gl/3cUF3ZVc7qy8uxAi9)
+
fatboyslim
Who else still doesn't know what T21 pills are and feels left out?
+
Help your fellow humans! (see more)
∗gigantichawk asks
(
step2ck_free120):
gigantichawk
"Septic arthritis may be definitively established in the setting of positive synovial fluid Gram stain/culture. In patients with purulent synovial fluid (leukocyte count 50,000 - 150,000) but negative synovial fluid cultures, a presumptive diagnosis of septic arthritis can be made."
Acute Calcium Pyrophosphate deposition would typically have a leukocyte count of 15,000 - 30,000 on aspiration.
+
∗azibird asks
(
step2ck_form8):
sorrel1000
@azibird that confused me too. I was thinking sarcoidosis and then put constrictive pericarditis down. But now looking back, there wasn't enough support for sarcoidosis.
+1
yogurt-dimple asks
(
nbme25):
raspberry-muffin
In this question sounds like inherited paternally , but this muscle biopsy confirmed Mitochondrial inheritance. Muscle biopsy: Immunohistochemistry typically shows ragged red fibers, which are caused by sub-sarcolemmal and inter-myofibrillar accumulation of defective mitochondria in muscles (mitochondria stain red). Probably Mother has the same condition too.
+
yogurt-dimple
Gotcha. Yeah, the red ragged fibers tipped me off to mitochondrial myopathy, but because the stem implied paternal inheritance, I figured there was just another disease I had forgotten about that presents with them.
+
drdoom
@raspberry-muffin I'm not convinced. It is highly unlikely the NBME would write this question and expect you to "assume" mom has condition without making any mention of mom. Plus, it is simply highly improbable that myopathy is present in both mom and dad lineage. That seems off to me.
+
drdoom
@yogurt-dimple, I think this a key line in the explanation: “However, there are additional mutations that affect mitochondrial RNA translation, trafficking and incorporation of respiratory protein complexes, and maintenance of the inner mitochondrial membrane that can also lead to mitochondrial myopathy.”
+
drdoom
Yes, they say, "Mitochondrial diseases are strictly inherited through the mother" but this is not a mitochondrial disease — this is a "non-mitochondrial–derived" mitochondrial myopathy; yes, mitochondria are affected but the mutation is in somatic (nuclear) genes that govern the maintenance of "healthy mitochondria". This is because the mutation affects the function/operation of mitochondria but the mutation itself is in the nuclear DNA (which control something about the "quality" of mitochondria but what exactly is not yet known).
+
∗kms123 asks
(
free120):
fhegedus
Eosinophils (FA 2020 page 408) are involved in type I hypersensitivity reactions (asthma, allergy, analphylaxis), parasitic infections and other pathologies. They are not involved in edema formation. I hope this helps! :)
+
fhegedus
Also, the patient in the question got a laceration, which probably led to a bacterial infection; so neutrophils would be predominant, not eosinophils.
+
∗kms123 asks
(
nbme22):
sd22
PT, PRT, and TT normal in antithrombin deficiency. FA ‘20 pg. 428
+
sd22
PTT* lol autocorrect clearly hasn’t been studying
+
Some shelves are here, too!
=>
Medicine 4
=>
Family Medicine 2
=>
Family Medicine 1
{kind=link}
{kind=link}
https://www.youtube.com/watch?v=4-DuvwoH2zQ if ur lazy like me, this is a good refresher video